GLI ANESTETICI LOCALI
Amedeo Pignataro
Servizio di anestesia e rianimazione, Ospedale Buccheri
La Ferla FBF, Palermo
1. Introduzione
Gli anestetici locali (AL) sono una categoria di farmaci il cui uso clinico
è in continuo crescendo; dalla scoperta nel 1884 della cocaina alla nuova
ropivacaina, gli anestetici locali determinano lo straordinario effetto di
“addormentare” una parte del corpo senza provocare la perdita di coscienza dell’anestesia
generale.
Il termine di anestetici locali comprende una classe
farmacologica eterogenea accomunata da un analogo meccanismo d'azione basato
sull'interruzione transitoria e reversibile della conduzione nervosa in
corrispondenza del sito in cui vengono applicati. Tra le numerose sostanze
dotate di questa proprietà, caratteristiche fisico-chimiche diverse e altri
fattori ne condizionano l'attività clinica e la tossicità.
In relazione alla struttura chimica, gli AL possono essere classificati in due categorie:
| Amino-esteri: procaina, clorprocaina, tetracaina | |
| Amino-amidi: lidocaina, mepivacaina, prilocaina, bupivacaina, etidocaina, ropivacaina |
Nonostante la loro eterogeneità, nella molecola della maggior parte degli AL
possono essere individuate tre porzioni costituite da (figura 1):
un polo lipofilo, rappresentato spesso da un anello
aromatico responsabile della liposolubilità del prodotto, della diffusione nei tessuti
e nelle membrane biologiche, del fissaggio alle proteine plasmatiche e
dell'attività.
un polo
idrofilo, comune alle due classi di AL, che conferisce agli
anestetici locali il carattere di amine terziarie e ne determina il carattere
di base. L'idrofilia condiziona l'idrosolubilità, quindi la diffusione della
forma non ionizzata, e la ionizzazione (che, ad un determinato pH, è funzione
del pKa) della molecola dell'anestetico locale.
una catena
intermedia, che per la presenza di un legame amidico o estereo
permette la classificazione degli AL.
|
|
|
Figura
1 |
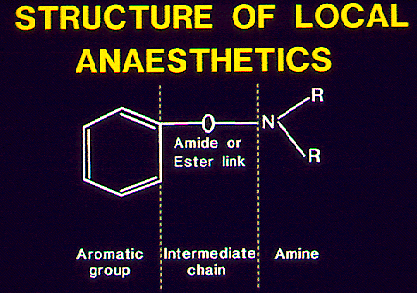
La natura della catena intermedia condiziona il metabolismo di queste
sostanze:
gli anestetici a
legame estereo sono facilmente e rapidamente idrolizzati nel plasma ad opera
delle pseudocolinesterasi.
gli anestetici
amidici vengono degradati meno rapidamente e catabolizzati pressoché unicamente
a livello dei microsomi epatici. Questo conferisce a queste molecole una
stabilità e una durata d'azione maggiore.
La lunghezza e le ramificazioni della catena intermedia determinano
inoltre, l'attività dell'AL.
In soluzione gli AL esistono in due forme:
1.
forma basica (R-NH2), non ionizzata e
liposolubile
2.
forma ionizzata [(R-NH3)+Cl-],
idrosolubile;
la proporzione tra le due forme dipende dalla costante di dissociazione
(Ka) della forma ionizzata e dal pH:
Ka = [H+] ][base]/[ac.
coniugato]
questa formula può essere riscritta secondo l'equazione di Henderson-Hasselbach:
pKa = pH+ log
base/ac.coniugato.
Per un pKa uguale al pH il rapporto tra le 2 forme sarà uguale. Al crescere
del pK prevarrà in soluzione la quota ionizzata.
Gli AL bloccano (in modo transitorio e reversibile) la conduzione nervosa
modificando la propagazione del potenziale d'azione a livello dell’assone. La
membrana della fibra nervosa, da una condizione di riposo mantenuta
dall'attività di una pompa Na-K ATPasi -dipendente, in seguito alla corrente
del potenziale d'azione, consente l'ingresso massivo di ioni sodio al suo
interno, modificando il potenziale di membrana da negativo a positivo. La
corrente di depolarizzazione nel momento in cui tutta la superficie della
membrana è depolarizzata, innesca modificazioni strutturali del canale sodico
responsabili di un ostacolo ad un'ulteriore ingresso di Na con conseguente
inattivazione dell'eccitabilità di membrana. All’interruzione della corrente
sodica segue la fuoriuscita dalla cellula di ioni K in numero uguale a quello
di ioni Na penetrati nella fase di depolarizzazione.
Il canale rapido del sodio è il recettore specifico su cui agiscono gli AL.
Le molecole d'anestetico, una volta attraversata la membrana cellulare della
fibra nervosa, si legherebbero ad un recettore presente sulla faccia interna
della membrana, di fatto impedendo l'ingresso massivo di ioni Na e quindi la
fase di depolarizzazione (figura 2).
|
|
| Figura 2 |

|
|
|
|
|
Filmato in
Real Player che
mostra l’interruzione dell’impulso nervoso lungo l’assone dopo la
somministrazione di AL. Click su l'immagine per visualizzarlo |
|
|
Gli AL nella loro forma attiva, quella ionizzata, interferiscono con le
diverse fasi del potenziale d'azione, diminuendone l'ampiezza, la velocità di
depolarizzazione e la durata del periodo refrattario. La concentrazione di AL
necessaria per determinare il blocco della conduzione nervosa differisce per
ogni sostanza, pertanto la potenza di ciascun AL viene indicata dalla
concentrazione minima inibente (Cmi), ovvero dalla concentrazione di farmaco al
di sotto della quale la fibra ritorna ad essere eccitabile.
4. Caratteristiche fisico-chimiche
La liposolubilità, il legame proteico e il pKa rappresentano fattori
farmacologici fondamentali di ciascun AL .
a. Liposolubilità.
La liposolubilità condiziona la potenza degli AL. Gli anestetici a maggiore
liposolubilità sono anche i più potenti; ciò dipende dalla capacità di
attraversare la matrice fosfolipidica delle membrane cellulari.
b. Legame proteico.
Ha un effetto significativo sulla durata del blocco. Una grossa percentuale
di AL legata alle proteine è l'espressione di una grande affinità di legame per
le proteine recettoriali. Gli AL a maggiore potenza hanno anche la maggiore
durata d'azione.
c. pKa.
Il pKa è definito come il pH al quale il 50% di un AL è presente in forma
ionizzata ed il 50% in quella non-ionizzata. Il pKa è importante nel
determinare l'onset di un AL. Anestetici con un pKa prossimo al pH fisiologico
avranno un onset più rapido perché presenti in soluzione in maggior parte nella
quota non-ionizzata (base). Al crescere del pK prevarrà in soluzione la quota
ionizzata responsabile di un onset lento. La forma basica, liposolubile, è
responsabile dell'interazione con i componenti del doppio strato lipidico
cellulare e condiziona la diffusione perinervosa, mentre la quota ionizzata
idrofila, dopo l'ingresso cellulare dell'AL, ne determina l'effetto
farmacologico.
5. Caratteristiche non fisico-chimiche
L'attività clinica degli AL può differire da quella in vitro, basata sulle diverse
proprietà fisico-chimiche delle molecole. Altri fattori, infatti, intervengono
in vivo (Tabella 1):
|
a |
|
|
Diffusibilità tessutale |
|
b |
|
|
Vasodilatazione |
|
c |
|
|
Metabolismo |
|
d |
|
|
Tipi di fibre |
|
e |
|
|
Tipo di blocco |
|
f. |
|
|
Aggiunta di un vasocostrittore |
Tabella 1
Tipi di fibre
Esiste una differente "sensibilità" delle fibre nervose agli AL:
il diametro e il grado di mielinizzazione delle fibre nervose sono i fattori ritenuti
maggiormente responsabili (tabella 2).
|
Ordine di blocco delle fibre nervose per gli anestetici
locali |
||||||
|
|
Aa Motoria |
Ab Epicritica |
Ag Propriocet. |
Ad Termoalg. |
B
Pregamgl. simpatica |
C
Termoalg. |
|
Mieliniz. |
4+ |
3+ |
2+ |
+ |
+ |
0 |
|
Diametro |
12-30
|
5-12 |
5-10 |
1-4 |
1-3 |
0,5-1 |
|
Ordine
blocco |
5 |
4 |
3 |
2 |
1 |
2 |
|
|
|
|
|
|
|
|
Tabella 2
La classica teoria di Gasser e Erlanger (1929) secondo la quale il diametro
della fibra è l'unico fattore che condiziona la sensibilità all'AL è stata
modificata, con la dimostrazione (in vitro) che, nelle fibre mieliniche i
fattori determinanti del blocco nervoso sono dati dalla concentrazione critica
dell'AL e dalla lunghezza della fibra bagnata dall'AL. Il numero minimo di nodi
di Ranvier, infatti, che deve essere bagnato dall'AL per determinare un blocco
dell’impulso nervoso è tre, indipendentemente dal diametro della fibra. La
distanza internodale e quindi il numero dei nodi varia tra fibre spesse e
sottili conducendo ad una diversa sensibilità all'AL delle fibre nervose.
la lidocaina,
per esempio, utilizzata per via peridurale alla concentrazione di 0.5% induce
un blocco simpatico, alla concentrazione dell'1% un blocco simpatico e
sensitivo e al 2% un blocco completo (simpatico, sensitivo e motorio).
la bupivacaina
ha, fino ad una certa concentrazione, una diffusione difficoltosa nelle fibre
di grosso calibro. L'aumento della concentrazione della soluzione di
bupivacaina permette di ottenere un blocco motore e riduce la velocità
differenziale del blocco.
l'etidocaina,
per contro, non presenta velocità differenziale del blocco ; essa blocca
facilmente le fibre di grosso calibro probabilmente per via della sua elevata
liposolubulità e del pKa minore di quello della bupivacaina. Per via
peridurale, perciò, il blocco motorio dell'etidocaina all'1% è maggiore e
s'instaura più rapidamente di quello della bupivacaina allo 0.25% o allo 0.50%,
mentre il blocco sensitivo della bupivacaina è quasi completo (95%) alla
concentrazione dello 0.25%. Una soluzione di etidocaina all'1.5% non induce un
blocco sensitivo come quello della bupivacaina allo 0.5%.
Sulle fibre C, gli anestetici locali agiscono tutti nella stessa maniera
determinando a dose equi-anestetica un blocco simpatico della medesima
intensità.
Tipi di blocco
La tabella 3 mette a confronto l'onset dell'effetto massimale e la durata
d'azione di tre anestetici locali per un blocco plessico e per un'anestesia
peridurale:
Tra i due tipi di blocco esistono delle differenze:
-
l'anestesia ottenuta con un blocco plessico è più
prolungata di quella peridurale per tutti gli AL
-
l'onset degli anestetici locali è più omogeneo
nell'anestesia peridurale che in quella plessica.
Tabella
3 - Onset e durata d'azione di alcuni anestetici locali per un blocco plessico
e per un'anestesia peridurale
|
TECNICA
ANESTETICA |
ANESTETICO |
C%
|
ONSET
MEDIO |
DURATA
MEDIA |
|
|
||||
|
Blocco
pl.brachiale |
Lidocaina |
1 |
14.04 ±
3.83 |
195 ±
26.3 |
|
Anestesia
peridurale |
Lidocaina |
2 |
15 |
100 ± 20 |
La tabella 4 confronta i tempi d'azione dell'anestesia peridurale e
subaracnoidea:
- la durata dell'effetto anestetico è minore per via subaracnoidea a causa
delle piccole dosi somministrate
- l'onset è più rapido per via subaracnoidea.
Le differenze dei tempi d'azione dei diversi anestetici locali sono
identiche per via peridurale e subaracnoidea.
La bupivacaina iperbarica alla concentrazione dello 0.5%-1% può essere
utilizzata, per via subaracnoidea, in dosi variabili da 7.5 a 25 mg.
Tabella
4 - Onset e durata d'azione degli anestetici locali per via peridurale e
subaracnoidea
|
|
|
C% |
DOSE
(mg) |
ONSET
(min) |
DURATA
(min) |
|
|
|||||
Anest.perid
|
Lidocaina |
1 .2 |
100-300 |
10-20 |
45-120 |
Anest.subaracnoidea
|
Lidocaina |
5 |
100 |
3 |
90 |
Aggiunta di un vasocostrittore
Per la maggior parte degli AL somministrati per via peridurale e nei
blocchi periferici, l'associazione di un vasocostrittore riduce il
riassorbimento ematico, aumenta il fissaggio degli anestetici locali sulla
fibra nervosa, migliorando la qualità e prolungando la durata del blocco.
Tra i vasocostrittori, l'adrenalina, alla concentrazione di 1/200000, è la
più efficace e la meno nociva; una concentrazione superiore (1/80000) non
procura alcun vantaggio supplementare e espone ad effetti sistemici e locali
d'ischemia.
L'effetto del vasocostrittore dipende, tuttavia, dall'anestetico locale e
dal sito di somministrazione:
- nei blocchi nervosi periferici, l'adrenalina prolunga l'azione di tutti
gli AL.
- per via peridurale, l'adrenalina aumenta la durata d'azione degli
anestetici locali a debole o intermedia attività intrinseca e interviene poco
per un anestetico a forte attività intrinseca.
Nel caso della bupivacaina, tuttavia, sembra che l'associazione con
l'adrenalina nell’anestesia subaracnoidea migliori la qualità del blocco (in
particolare di quello motorio) ed aumenta la durata dell'anestesia.
6. Tossicità acuta

A differenza degli altri farmaci, la concentrazione ematica degli AL è un
effetto indesiderabile che può essere responsabile d'effetti tossici.
I fattori che influenzano la tossicità degli AL sono elencati in tabella 5:
|
A |
le proprietà fisico-chimiche: queste rendono conto della tossicità propria di ogni sostanza |
|
B |
i fattori che modificano il tasso plasmatico, poiché
la tossicità acuta di un anestetico locale è condizionata da: |
|
C |
i fattori suscettibili di ridurre la soglia di tossicità neurologica e cardiovascolare dei differenti AL. |
Tabella 5
Tossicità neurologica
Il primo segno di tossicità sistemica degli AL è la sonnolenza o un senso
di ebbrezza; seguono parestesia nella regione circumorale, sensazione di lingua
addormentata, tinniti, disturbi visivi, agitazione. In intossicazioni severe si
manifestano convulsioni, coma e depressione cardiorespiratoria. La potenza
d'azione è un fattore importante della tossicità generale degli AL: esiste,
infatti una relazione tra efficacia e tossicità neurologica: gli AL più
efficaci sono anche i più tossici ); questo dipende dal grado di
liposolubilità: più il farmaco è liposolubile meglio attraversa la barriera
emato-encefalica. La tossicità potenziale di una sostanza viene quantificata
dalla sua soglia plasmatica, che corrisponde alla concentrazione venosa alla
quale compaiono le prime manifestazioni neurologiche.
La lidocaina e la mepivacaina hanno la stessa soglia di tossicità. La
bupivacaina e l'etidocaina sono i più tossici tra gli AL utilizzati in clinica:
per la bupivacaina i primi segni di tossicità possono comparire a partire da
1.6 mcg/ml, le convulsioni da 4 mcg/ml (tabella 6). La soglia plasmatica di
tossicità neurologica diminuisce nelle stesse proporzioni dell'aumento della
potenza d'azione.
|
|
ATTIVITA' |
TOSSICITA |
C TOSSICA |
|
POTENZA
INTERMEDIA |
|
|
|
|
Mepivacaina |
2 |
1.9 |
5-6 |
|
Lidocaina |
|
|
5-6 |
|
POTENZA
ELEVATA |
|
|
|
|
Bupivacaina |
16 |
6 |
1.6 |
|
Etidocaina |
16 |
5 |
2 |
Tabella 6
Tossicità cardiaca
Alla somministrazione endovenosa accidentale di 100-200 mg di un potente
anestetico locale (es. Bupivacaina), si assiste nell’uomo alla comparsa di
turbe del ritmo, come extrasistoli ventricolari, tachicardie ventricolari e
sopraventricolari e di difetti di conduzione con allargamento del QRS. La
velocità con la quale si raggiunge la concentrazione plasmatica massimale (T
Max) è un fattore determinante nella tossicità di un AL: un'eccellente
prevenzione di questi problemi consiste, per via peridurale, nell'iniezione
lenta e frazionata (5 ml/min) della soluzione dell'AL, mentre l'utilizzazione
di una dose test con aggiunta di adrenalina, soprattutto nelle donne in travaglio,
è controversa e non consigliabile.
Nonostante le dimostrazioni in vitro della cardioselettività di alcuni AL
(es. lidocaina e mepivacaina) per cui le convulsioni precedono sempre i segni
di tossicità cardiaca (soglia plasmatica di tossicità cardiaca 2-3 volte
superiore di quella convulsivante), questi dati non sono stati ritrovati in
studi animali nei quali il sistema cardiovascolare è risultato sempre più
resistente agli AL del SNC, per tutti gli anestetici testati.
Per gli AL con potenza maggiore, l'aumento della tossicità non è
perfettamente correlata ad un aumento dell'attività anestetica: la bupivacaina
è stimata 17 volte più cardiotossica della lidocaina, mentre possiede
un'attività anestetica soltanto 4 volte maggiore di quest'ultima.. La
bupivacaina ha, in effetti, un'azione aritmogena maggiore di quella della
lidocaina. Quest'ultima è antiaritmogena a dosi non tossiche, mentre la
bupivacaina e l'etidocaina, qualunque sia la dose, inducono un effetto
stabilizzante di membrana procainamide-simile a livello cardiaco. Quest'azione
è aritmogena per piccoli aumenti della dose; ciò spiega la possibilità di
trattare le turbe dell'eccitabilità miocardica ventricolare indotte dalla
bupivacaina con la somministrazione di lidocaina.
Fattori che potenziano la tossicità cardiaca, comuni a tutti gli AL che ne riducono il margine di sicurezza sono mostrati in tabella 7:
| L'acidosi aumenta la captazione cellulare a livello polmonare, renale, epatico e cardiaco soprattutto degli AL con pKa elevato | |
| L’iperKaliemia: favorisce le turbe della conduzione indotte dagli AL | |
| ipossia miocardica. |
Per gli AL più potenti, bisogna aggiungere un fattore favorente
supplementare rappresentato dalle condizioni cardiache preesistenti che, quindi,
pur non controindicandone l'uso, richiedono particolare attenzione nel calcolo
del dosaggio.
Tossicità
fetale
La tossicità fetale si traduce clinicamente in convulsioni intermittenti,
bradicardia o apnea. Si manifesta essenzialmente nel caso di fattori favorenti
associati come, per esempio, un'acidosi respiratoria o metabolica o un'anossia,
tutti elementi che riducono la soglia convulsiva e aumentano il passaggio
placentare del farmaco. Gli AL (esteri o amidi), una volta raggiunta la
circolazione sistemica, attraversano più o meno tutti la barriera placentare
per via della loro liposolubilità e del peso molecolare relativamente basso. Il
trasporto placentare è determinato soprattutto dalla quota di anestetico legato
alle proteine plasmatiche: soltanto la forma libera, non fissata alle proteine
plasmatiche, attraversa la barriera placentare; della forma libera, il
passaggio interessa esclusivamente la parte non ionizzata, ed è perciò
dipendente dal pKa. Maggiore, quindi, è la quota di anestetico locale fissata
alle proteine plasmatiche, più basso è il rapporto feto-materno (tabella 8).
Per la bupivacaina vi è una correlazione tra il rapporto delle concentrazioni
plasmatiche feto-materne e le variazioni individuali delle frazioni libere
dosate . Questo fenomeno è potenziato dalla carenza relativa, fisiologica nel
feto, di a1 globuline, che costituiscono la quota proteica che lega
maggiormente gli AL. La forma legata dell'AL si riduce ad 1/3 o 2/3 di quella
della madre e questo ne aumenta la quantità materna totale. Gli AL ad alta
affinità di legame con le proteine plasmatiche (bupivaciana, ropivacaina),
sono, quindi, da preferirsi in anestesia ostetrica.
Tabella 8 - Legame proteico e rapporto feto-materno
(F/M)
ANESTETICO
|
LEGAME PROTEICO % |
RAPPORTO F/M |
|
LIDOCAINA |
64 |
0.52-0.69 |
|
MEPIVACAINA |
77 |
0.69-0.71 |
|
BUPIVACAINA |
95 |
0.31-0.44 |
Gli AL subiscono, come qualsiasi sostanza introdotta in un organismo, un processo
di assorbimento, una fase di distribuzione, sono soggetti generalmente a
diversi sistemi di detossificazione (metabolismo) e finalmente vengono escreti
(eliminazione).
Il riassorbimento ematico degli AL dipende:
|
1 |
dal
sito d'iniezione |
|
2 |
dalla
dose |
|
3 |
dall'associazione
o meno ad un vasocostrittore |
|
4 |
dalle
proprietà fisico-chimiche di ciascun anestetico locale |
1. Sito di
somministrazione:
il riassorbimento ematico, in generale, dipende dalla ricchezza di vascolarizzazione
della regione anatomica e dalla presenza di tessuti e di grassi capaci di
fissare localmente gli AL, particolarmente quelli altamente liposolubili:
le regioni
intercostali e peridurali hanno una vascolarizzazione identica, ma la presenza di
grassi perimidollari giustifica il maggiore riassorbimento dell'anestetico per
via intercostale rispetto a quella peridurale .
Per via
subaracnoidea, 3 ml di bupivacaina allo 0.5 % inducono concentrazioni plasmatiche
molto piccole. La dura madre, tuttavia, come dimostrato per la lidocaina, non
riduce il riassorbimento locale dell'anestetico. La dura riduce, invece, la
velocità di riassorbimento: la T Max è maggiore per via subaracnoidea che per
via peridurale. L'elemento essenziale, in realtà, è la minore dose
somministrata per la prima via che per la seconda.
2. Dose somministrata:
per tutti gli AL, in genere, le Cp Max aumentano con il crescere della
dose. E' possibile, dunque, indicare le dosi massime da non superare alla prima
somministrazione (tab.5)
Tabella 9 - Dosi massime
iniziali di AL
ANESTETICO
|
C% |
TIPO
DI BLOCCO |
DOSE
MAX INIZIALE (mg) |
|
LIDOCAINA |
0.5 |
Endovenoso |
300 |
|
|
1-2 |
An. perid. |
500 + Adr |
|
|
5 |
IR |
100 |
|
MEPIVACAINA |
1 |
Endovenoso |
300 |
|
|
1.5-2 |
An.
perid. |
500 +
Adr |
|
BUPIVACAINA |
0.5-0.75 |
An.
perid. |
150 +
Adr |
|
|
|
|
|
3. Aggiunta di un vasocostrittore
A livello peridurale, l'associazione di un vasocostrittore ad alcuni AL (lidocaina)
permette di ridurre le Cp Max riducendo il riassorbimento ematico. L'aggiunta del vasocostrittore
ad altri AL (etidocaina, bupivacaina) ha scarso effetto sulle concentrazioni
plasmatiche: per l'etidocaina e la bupivacaina l'elevata liposolubilità rende
conto della captazione del grasso perimidollare. Il potenziale effetto
vasodilatatore di queste sostanze, inoltre, annulla quello vasocostrittore
dell'adrenalina. Per contro, a livello dei blocchi periferici l'adrenalina
riduce il riassorbimento ematico di tutti gli AL: l'uso d'una soluzione di
bupivacaina CON adrenalina per un
blocco del plesso brachiale per via sopraclavicolare riduce il valore della Cp
Max del 50%.
4. Proprietà fisico-chimiche dell'anestetico locale
La relazione dose somministrata/Cp Max, varia da un anestetico ad un altro,
e dipende dalla liposolubilità e dal metabolismo del prodotto. Per dosi
identiche, più l'AL è liposolubile minore è la Cp Max:
Metabolismo
I tassi ematici di un AL dipendono dal suo metabolismo: la prilocaina è
catabolizzata più rapidamente a livello epatico della lidocaina, quindi per una
stessa dose somministrata la Cp Max della prilocaina è minore di quella della
lidocaina.
Riassumendo possiamo dire che:
maggiore è la liposolubilità di un AL maggiore è la sua potenza; questa ne
riduce la soglia di tossicità neurologica e il riassorbimento ematico nello
spazio peridurale. Per la maggior parte degli AL, d'altronde, il rapporto C
tossica/Cp Max è lo stesso (tabella 10).
Tabella 10 - Cp Max e
rapporto tra concentrazione tossica (C Tox) e Cp Max degli AL somministrati per
via peridurale
|
|
Cp MAX |
C Tox/Cp Max |
DOSE |
|
LIDOCAINA |
1-2% |
2.4 2.3 |
400 + Adr |
|
MEPIVACAINA |
1.5-2% |
2.8 2 |
400 + Adr |
|
BUPIVACAINA |
0.5% |
0.6 2.7 |
100 + Adr |
pH plasmatico
Il pH plasmatico influenza numerosi fattori. L'acidosi infatti:
|
|
|
|
|
|
2 Parametri farmacocinetici
Lo studio delle concentrazioni plasmatiche degli AL comprende anche quello
dei parametri farmacocinetici. Tra questi la conoscenza dell'emivita d'eliminazione
consente di prevenire, tenuto conto della durata di ciascuno, il rischio
eventuale d'accumulo in caso di reiniezione. Tali parametri sono stati
calcolati da Tucker e Scott a partire dalla cinetica plasmatica ottenuta dopo
iniezione ev in bolo dei differenti AL (tabella 11).
Tabella 11 - Parametri
farmacocinetici dopo somministrazione endovenosa
|
|
LIDOCAINA
|
MEPIVACAINA |
BUPIVACAINA |
ETIDOCAINA |
|
VD (L) |
91 |
84 |
209 |
666 |
|
T 1/2 p (H) |
0.016 |
0.012 |
0.045 |
0.036 |
|
T 1/2 a (H) |
0.16 |
0.12 |
0.46 |
0.31 |
|
T 1/2 b (H) |
1.6 |
1.9 |
3.5 |
2.6 |
|
Cl
(l/min) |
0.95 |
0.78 |
0.47 |
1.22 |
|
EE^ |
0.65 |
0.52 |
0.38 |
0.74 |
|
FL^ |
0.36 |
0.22 |
0.07 |
0.09 |
_________________________________________________________________
^ EE=
estrazione epatica FL= frazione libera
Lidocaina
Per evitare la comparsa di fenomeni d'accumulo conviene aggiungere un terzo
della metà della dose iniziale ogni 2 ore.
Bupivacaina e Etidocaina
L'emivita d'eliminazione della bupivacaina è maggiore di quella
dell'etidocaina benché quest'ultima abbia, per via della sua liposolubilità, un
volume di distribuzione maggiore. La degradazione epatica dell'etidocaina è più
rapida di quella della bupivacaina [63]. Nel caso della bupivacaina la
reiniezione di metà dose iniziale ad intervalli di 3 ore e mezzo non aumenta la
Cp Max. Benché, quindi, gli AL con maggiore potenza abbiano dei t1/2 b più
elevati di quelli ad attività intermedia, la loro maggiore durata d'azione
permette di distanziare le somministrazioni riducendo il rischio d'accumulo.
Tossicità locale
La neurotossicità per applicazione locale degli AL attualmente utilizzati
è, in principio, nulla perché le concentrazioni abituali sono di gran lunga
inferiori a quelle tossiche.
CONCLUSIONI
Gli AL presentano proprietà farmacologiche diverse in funzione delle loro
caratteristiche fisico-chimiche e del metabolismo:
Tra gli AL a potenza media:
- lidocaina e mepivacaina:
Per tutti gli AL di questa classe, esiste un ampio margine di sicurezza
della tossicità neurologica e cardiaca.
Nel gruppo degli AL più potenti:
poiché la liposolubilità condiziona il legame proteico, potenza e durata
d'azione sono indissociabili
- l'etidocaina induce un blocco motore predominante, spiegabile con la sua
elevata liposolubilità
- la bupivacaina, per il suo pKa elevato, crea un blocco differenziale tra
la soluzione a 0.25% e quella a 0.50%.
8. Ropivacaina
La Ropivacaina è un nuovo anestetico locale di tipo amidico con struttura
chimica simile a quella della bupivacaina e della mepivacaina. E’ preparato
come un s-isomero piuttosto che come una miscela racemica come la bupivacaina.
L’uso di s-isomeri piuttosto che di miscele racemiche in genere riduce la
tossicità sistemica di una molecola. Tra le proprietà fisico-chimiche, il pKa
(8) e il legame proteico (95%) sono comparabili a quelle della bupivacaina.
L’allarmante segnalazione di casi di arresto cardiaco durante anestesia
regionale associato all’uso di anestetici come bupivacaina e etidocaina ha
spinto verso la ricerca e sintesi di un nuovo anestetico locale .
La ropivacaina, recentemente introdotta in commercio anche in Italia,
sembrerebbe, in vitro, dotata di minori effetti negativi sull'elettrofisiologia
cardiaca e sul SNC. I risultati di uno studio condotto su un preparato
muscolare di fibre di Purkinje, hanno mostrato che la bupivacaina causa la
maggiore depressione sull’eccitabilità e sulla conduzione cardiaca, la
lidocaina ha il minore effetto depressivo, mentre la ropivacaina occupa una
posizione intermedia tra i due anestetici. Analogo comportamento si verifica
sull’attività elettrica cerebrale in seguito alla somministrazione endovenosa
in ratti, cani e conigli, di bupivacaina, lidocaina e ropivacaina. La dose
convulsivante della ropivacaina era maggiore della bupivacaina, ma inferiore a
quella della lidocaina. La ropivacaina attraversa rapidamente la
placenta : il minore legame proteico dell’anestetico nel feto riguardo
alla madre, condiziona nel feto una concentrazione plasmatica minore di quella
materna .
Clinicamente la ropivacaina è simile alla bupivacaina ma mostra alcune
differenze che, probabilmente la renderanno il farmaco di scelta in anestesia
regionale.
Paragonata alla bupivacaina, la ropivacaina, infatti, possiede (alla
stessa concentrazione sierica):
|
|
|
|
|
|
|
|
|
|
La ropivacaina, quindi, sarebbe dotata di
un blocco differenziale (blocco sensitivo > blocco motorio) maggiore della
bupivacaina.
Una proprietà interessante della ropivacaina è quella per cui, diversamente
da altri anestetici locali, il farmaco ha minime proprietà vasodilatatrici.
L’aggiunta di epinefrina, pertanto, non influenza significativamente la durata
e la qualità del blocco e non incide sul livello plasmatico del farmaco quando
usato in anestesia regionale. Lavori condotti utilizzando ropivacaina in infusione continua nel periodo
postoperatorio hanno mostrato, anche in questo contesto, una maggiore
selettività del blocco sensitivo in rapporto a quello motorio e una clearance
maggiore rispetto alla bupivacaina.
La ropivacaina (0,2%) e la bupivacaina (0,25%) sono state valutate in
infusione continua alla velocità di 6-8 ml/h in analgesia peridurale nel
travaglio di parto: entrambi i farmaci hanno prodotto un effetto analgesico
efficace e comparabile senza effetti negativi sull’outcome fetale. Nel taglio
cesareo condotto in anestesia peridurale, bupivacaina e ropivacaina alla concentrazione
di 0,50% hanno prodotto un’adeguata ed equivalente anestesia chirurgica, anche
se la ropivacaina ha mostrato un onset e una durata del blocco motorio minore.
Sebbene siano necessari ulteriori studi, la ropivacaina si propone come una
buona alternativa alla bupivacaina in ostetricia.
9. Anestetici locali in ostetricia
Gli AL sono diffusamente utilizzati in ostetricia a scopo analgesico
durante il travaglio e nel taglio cesareo.
Secondo Bromage, i requisiti ideali di un AL per l'analgesia peridurale
durante il travaglio ed il parto sono:
|
a |
Analgesia efficace e pilotabile |
|
b |
Sicurezza per la madre |
|
c |
Nessuno o minimo indebolimento della forza muscolare |
|
d |
Nessuna alterazione sulla progressione del feto |
|
e |
Nessuna depressione del feto |
Ciascuno dei 5 punti può essere raggiunto con la scelta dell'AL più
indicato, alla dose e alla diluizione idonea, considerando che l'analgesia
richiesta per il parto vaginale è inferiore a quella necessaria per un intervento
chirurgico; l'eventualità che dopo un travaglio in AP si debba ricorrere
all'applicazione di un forcipe o al taglio cesareo non implica, tuttavia, il
ricorso ad altri farmaci ne tantomeno all'anestesia generale. Le anestesie
rachidee costituiscono in ostetricia le tecniche più indicate sia
nell'analgesia del travaglio che nel taglio cesareo, mentre l'anestesia
generale trova oggi indicazione soltanto in particolari situazioni (distacco di
placenta, prolasso di funicolo).
Tra gli AL disponibili in clinica, la bupivacaina è considerato fino ad
oggi il farmaco di scelta in ostetricia. Il maggiore vantaggio di questo
anestetico è la durata d'azione prolungata (2-3 volte maggiore della
lidocaina); la bupivacaina è estremamente potente. Anche se possiede un onset
elevato (20 min.) dovuto ad un elevato pKa (8.1) che condiziona in vivo una
predominanza della forma ionizzata, meno diffusibile, rispetto alla quota
indissociata, liposolubile, le particolari caratteristiche anatomiche e
ormonali della gravida, lo riducono a circa 10 minuti. L'ottima qualità
dell'analgesia in relazione al blocco motorio, ovvero la possibilità,
particolarmente evidente alle più basse concentrazioni d'anestetico, di
ottenere un'analgesia senza effetti negativi sulla forza muscolare e l'effetto
prolungato, rendono la bupivacaina un eccellente anestetico nell'analgesia
peridurale in ostetricia in boli e/o in infusione continua. Pur attraversando
la barriera placentare, il trasferimento è limitato dalla grossa quota di
farmaco legata alle proteine plasmatiche materne, senza pertanto provocare
concentrazioni tossiche nel feto. La bupivacaina iperbarica all'1%, ad una dose
approssimativamente ridotta di un terzo rispetto alle donne non gravide,
produce un'ottima analgesia e un profondo blocco muscolare ideale per il taglio
cesareo.
La lidocaina, per molti anni l'anestetico standard per il travaglio in
peridurale, produce una buona analgesia con un onset minore della bupivacaina.
La sua durata d'azione relativamente breve, se comparata con quella della
bupivacaina, e, soprattutto, alcune segnalazioni di effetti depressivi fetali,
ne avevano messo in dubbio la sicurezza e limitato l'uso nell'analgesia
ostetrica. Studi seguenti che ne hanno riaffermato la sicurezza e
l'affidabilità, hanno permesso di riannoverarlo nel bagaglio farmacologico
dell'analgesia peridurale per il travaglio ed il parto. Viene abitualmente
utilizzata in miscela con la bupivacaina per abbreviare l'installazione del
blocco e sul piano perineale in fase espulsiva o subito dopo il parto per la
riparazione chirurgica del piano perineale. La lidocaina, inoltre, è
considerato ancora il migliore anestetico per la dose test nel corso
dell’anestesia peridurale per via dekl suo rapido onset e bassa tossicità
cardiaca e neurologica rispetto alla bupivacaina.
La ropivacaina sta rapidamente affiancandosi nell’uso clinico alla
bupivacaina anche in campo ostetrico: le due molecole, malgrado una stechiometria
ed una liposolubilità differente, presentano analogo comportamento nell’attraversamento
della placenta a causa del basso rapporto feto-materno dei due anestetici e delle
variazioni del pH fetale, che riducendosi, in condizioni di acidosi, provoca un
aumento del trasferimento placentare degli anestetici.
10. Oppioidi
Gli effetti clinici degli oppioidi vengono sfruttati in terapia da secoli.
Il loro uso parenterale in anestesia, nel periodo postoperatorio e nel
trattamento del dolore acuto e cronico, si avvale di molte molecole con
caratteristiche fisico-chimiche e farmacocinetiche differenti. Dall'epoca in
cui (1970) fu dimostrato per la prima volta, che gli oppioidi somministrati per
via midollare, producevano una buona analgesia, innumerevoli studi ed
esperienze cliniche ne hanno confermato l'efficacia, proponendo gli oppioidi
come farmaci sicuri anche nell'analgesia peridurale in ostetricia. La scoperta
e la localizzazione dei recettori degli oppiacei a livello midollare e in molte
altre aree del SNC, alimentata da lavori sperimentali sulla distribuzione dei
derivati morfinici iniettati intratecalmente, ha indotto a ritenere questa via
di somministrazione più sicura in termini di riduzione della dose efficace e
degli effetti collaterali, rispetto alla via parenterale. La morfina, per via
della sua idrosolubilità che ne permette una significativa riduzione della
dose, può essere utilizzata per via rachidea; gli oppioidi più solubili, da
soli per via intratecale nel trattamento del dolore acuto non hanno vantaggi
rilevanti rispetto all'iniezione parenterale, se non nella possibilità per il
fentanyl di una riduzione del 25% della dose. In ogni caso il rischio d'effetti
collaterali limita l'uso degli oppioidi ad ambienti dotati di adeguato
controllo medico-infermieristico. Nell'analgesia peridurale in ostetricia, gli
oppioidi da soli, pur determinando un'analgesia soddisfacente durante la I fase
del travaglio, non garantiscono un buon sollievo del dolore nel periodo
espulsivo. A parte la depressione respiratoria, anche gli effetti collaterali
minori (nausea, prurito, disuria) degli oppioidi, non vengono accettati
facilmente dalla donna gravida.
L'associazione di piccole dosi di oppioidi agli AL per via peridurale,
invece, è una scelta interessante per diversi motivi:
|
a) riduzione del dosaggio degli AL |
|
b) riduzione dell'onset del blocco |
|
c) minore blocco motorio |
|
d) maggiore comfort per la paziente |
Il razionale dell'associazione farmacologica è nell'effetto dei due farmaci
a due diversi livelli: l'AL agisce sull'assone del nervo e l'oppioide sui
recettori spinali producendo un effetto additivo di blocco e di modulazione
dello stimolo nocicettivo. Le modalità di somministrazione per il travaglio e
il parto vanno dal bolo di AL-oppioidi seguito da rifornimenti ad intervalli
costanti, al bolo misto seguito da un'infusione continua di AL+ oppioidi. I
farmaci più utilizzati sono la bupivacaina 0.25% + fentanyl 50 mcg per il bolo,
seguite da soluzioni (bupivacaina 0.25-0.125% + fentanyl 1-3 mcg/ml a velocità
variabile da 6-12 ml/h) in infusione continua. Nonostante numerosi studi documentino
l'efficacia in termini d'analgesia e l'assenza d'effetti negativi sulla madre e
sul feto dell'associazione AL-oppioidi in infusione continua durante il
travaglio, considerata l'impossibilità di prevedere il livello degli oppioidi
nel liquor e il raggiungimento dei centri sopraspinali dopo infusioni a volte
prolungate, la pratica del nostro servizio si limita all'aggiunta di piccole
dosi di fentanyl (50 mcg) all'AL nel bolo peridurale iniziale, proseguendo con
i soli AL l'infusione continua per il travaglio ed il parto. Nel taglio cesareo
d'elezione e in quello consecutivo ad un travaglio condotto in AP, il fentanyl
(1 mcg/Kg) può essere aggiunto alla bupivacaina (0.50%, 15-20 ml) per ridurre
l'onset del blocco e garantire una blanda sedazione alla donna gravida.
11. Coadiuvanti
La clonidina è un'agonista alfa 2-adrenergico da alcuni anni utilizzato in
anestesia generale per i suoi effetti sedativi, ansiolitici, analgesici, e di
stabilizzazione emodinamica; tali effetti sono dovuti ad un’azione sul SNC
(sedazione, ansiolisi, analgesia) e ad effetti centrali e periferici sul
sistema cardiovascolare (bradicardia, ipotensione).
L'efficacia della clonidina per via peridurale nel trattamento del dolore
postoperatorio e del dolore cronico, da solo o in associazione con gli oppioidi
è stato documentata in diversi lavori. Recentemente la clonidina è stata
impiegata (75 mcg) insieme alla bupivacaina e al fentanyl per l'AP per il
travaglio: l’associazione della clonidina agli anestetici locali, esercitando
un’azione sinergica, permetterebbe una riduzione del dosaggio degli agenti
anestetici. Considerando tuttavia l'esiguità delle segnalazioni in letteratura
sull'effetto materno-fetale del farmaco, l'uso della clonidina non può essere
raccomandato per la pratica clinica nell’analgesia peridurale in ostetricia.
BLOCK
A., COVINO B.G. Effect of local anaesthetic agents on cardiac conduction and
contractility.Reg. Anaesth. 6: 55-61, 1981
BROMAGE
P.R. Mechanism of action of extradural analgesia. Br. J. Anaesth. 47: 199-212,
1975
BROMAGE
P.R. Epidural analgesia. Saunders W.B., USA, 1978
COLLEY
P.S., HEAVNER J.E. Blood levels of bupivacaine after injection into the scalp
with and without epinephrine. Anesthesiology 58: 81-84, 1981
CONKLIN
K.A., ZIADLOU-RAD F. Bupivacaine cardiotoxicity in a pregnant patient with
mitral valve prolapse. Anesthesiology 58: 596, 1983
COVINO
B.G., VASSALO H.D. Local anaesthetics: mechanisms of actions and clinical use.
Grune and Stratton, New York, 1976
DATTA
S., BROWN W.U., OSTHEIMER G.W., WEISS J.B., ALPER M.H. Epidural anaesthesia for
cesarian section in diabetic parturients: maternal and neonatal acid-base
status and bupivacaine concentration. Anesth. Analg. 60: 574-578, 1981
de
JONG R.H., DAVIS N.L. Treating bupivacaine arrythmias preliminary report. Reg.
Anaesth. 5: 99-103, 1981
KUNHERT B.R., PHILIPSON E.H., PIMENTAL R., KUNHERT P.M. Assessment of motor
blockade during epidural anaesthesia. Anesthesiology 56: 477-478,1982
LANZ
E., THEISS D., KELLNER G., ZIMMER M., STAUDTE H.W. Assessment of motor blockade
during epidural anaesthesia. Anesth. Analg. 62: 889-893, 1983
LITTLEWOOD
D.G., BUCKLEY P., COVINO B.G., SCOTT D.B., WILSON J. Comparative study of
various local anaesthetic solution in extradural block in labour. J. Anaesth.
51: 475-515, 1979
LIU
P.L., FELDMAN H.S., COVINO B.G. Comparative CNS and cardiovascular toxicity of
various local anaesthetic agents. Anesthesiology 55: A 156, 1981
MATHER
L.E., TUCKER G.T. Pharmacokinetics and biotransformation of local anaesthetics
Int. Anaesthesiol. Clin. 16: 23-51, 1978
MOORE
D.C. et al. Arterial and venous plasma levels of bupivacaine following epidural
and intercostal nerve block. Anesthesiology 45: 39-45, 1976
PEDERSEN
H., MORISHIMA H.O., FINSTER M. Uptake and effects of local anaesthetics in
mother and fetus. Int. Anaesthesiol. Clin. 16: 73-89, 1978
PRENTISS
J.E. Cardiac arrest following caudal anaesthesia. Anesthesiology 50: 51-53,
1979
RALSTON
D.H., SHNIDER S.M. The fetal and neonatal effects of regional anaesthesia in
obstetrics. Anesthesiology 48: 34-54, 1978
REYNOLDS
F., HARGROVE R.L., WYMAN J.B. Maternal and foetal plasma concentrations of
bupivacaine after epidural block. Br. J. Anaesth. 45: 1049-1053, 1973
REYNOLDS
F., TAYLOR G. Plasma concentration of bupivacaine during continuous epidural
analgesia in labour: the effect of adrenaline. Br. J. Anaesth. 43: 436-440,
1971
SCANLON
J.W., OSTHEIMER G.W., LURIE A.O., BROWN W.U., WEISS J.B., ALPER M.H.
Neurobehavioral responses and drug concentrantions in newborns after maternal
epidural anaesthesia with bupivacaine. - Anesthesiology 45: 400-405, 1976
SCOTT
D.B. Toxicity caused by local anesthetic drugs. Br. J. Anaesth. 53: 553-554,
1981
STEEN
P.A., MICHENFELDER J.D. Neurotoxicity of anesthetics. Anesthesiology 50:
437-453, 1979
THOMAS
J., LONG G., MOORE G., MORGAN D. Plasma protein binding and placental transfer
of bupivacaine. Clin. Pharm. Therap. 19: 426-434, 1976
TUCKER
G.T., COOPER S., LITTLEWOOD D., BUCLEY F.P., COVINO B.G., SCOTT D.B. Observed
and predicted accumulation of local anaesthetic agents during continuous
extradural analgesia. Br. J. Anaesth. 49: 237-242, 1977
TUCKER
G.T., MATHER L.E. Pharmacokinetics of local anaesthetic agent. Br. J. Anaesth.
47: 213-278, 1975
TUCKER
G.T., MATHER L.E. Clinical pharmacokinetics of local anaesthetics. Clin.
Pharmacokin. 4: 241-278, 1979
WIDMAN
B. Plasma concentration of local anaesthetic agents in regard to absorption,
distribution and elimination, with special reference to bupivacaine. - Br. J.
Anaesth. 47: 231-236, 1975
McCLURE
J.H.:Ropivacaine Br. J. Anaesth. 76:300-307, 1996
EDDLESTON
J.M. et al.: A double-blind comparison of 0.25% ropivacaine and 0.25%
bupivacaine for extradural analgesia in labour. Br. J. Anaesth. 76:66-71, 1996
DATTA
S. et al.:Clinical effects and maternal anf fetal plasma concentrations of
epidural Ropivacaine versus Bupivacaine for Cesarian section. Anesth.
1346-1353, 1995